ELAPRASE® (idursulfase) was studied in a pivotal clinical trial involving Hunter syndrome patients aged 5 years and older. It was also studied in an open-label extension study and a safety study involving patients aged 7 years and younger.1
Pivotal trial design
In the pivotal trial, ELAPRASE achieved the primary endpoint, improved walking capacity, reduced urinary GAG levels, and reduced liver and spleen volume in patients ≥5 years old.1 Read the ELAPRASE Important Safety Information here.
PIVOTAL TRIAL DESIGN
- Safety and efficacy evaluated in a 53-week, randomized, double-blind, placebo-controlled clinical trial of 96 patients with Hunter syndrome, ages 5 to 31 years old.1
-
Patients received either:1,2

- Primary efficacy outcome assessment was a two-component composite score based on the sum of the ranks of the change from baseline to Week 53 in distance walked in 6 minutes (6-minute walk test [6-MWT]) and the ranks of the change in %-predicted forced vital capacity (%-FVC).1,2
- Pharmacodynamic assessments included urinary GAG levels and changes in liver and spleen volume.1
Pivotal trial safety profile
- Hypersensitivity reactions were reported in 69% (22 of 32) of patients in the ELAPRASE once-weekly group.1
Adverse reactions that occurred in at least three patients (≥9%) in the ELAPRASE once-weekly group and had a higher incidence than in the placebo group included:1
Adverse Reactions that Occurred in the Placebo-Controlled Trial in At Least 9% of Patients in the ELAPRASE 0.5 mg/kg Once Weekly Group and with a Higher Incidence than in the Placebo Group (5 Years and Older)
| System organ class adverse reaction |
ELAPRASE (0.5 mg/kg weekly) N=32 n (%) |
Placebo N=32 n (%) |
|---|---|---|
| Gastrointestinal disorder | ||
| Diarrhea | 3 (9%) | 1 (3%) |
| Musculoskeletal and connective tissue disorders |
||
| Musculoskeletal pain | 4 (13%) | 1 (3%) |
| Nervous system disorders | ||
| Headache | 9 (28%) | 8 (25%) |
| Respiratory, thoracic, and mediastinal disorders |
||
| Cough | 3 (9%) | 1 (3%) |
| Skin and subcutaneous tissue disorders |
||
| Pruritus | 8 (25%) | 3 (9%) |
| Urticaria | 5 (16%) | 0 (0%) |
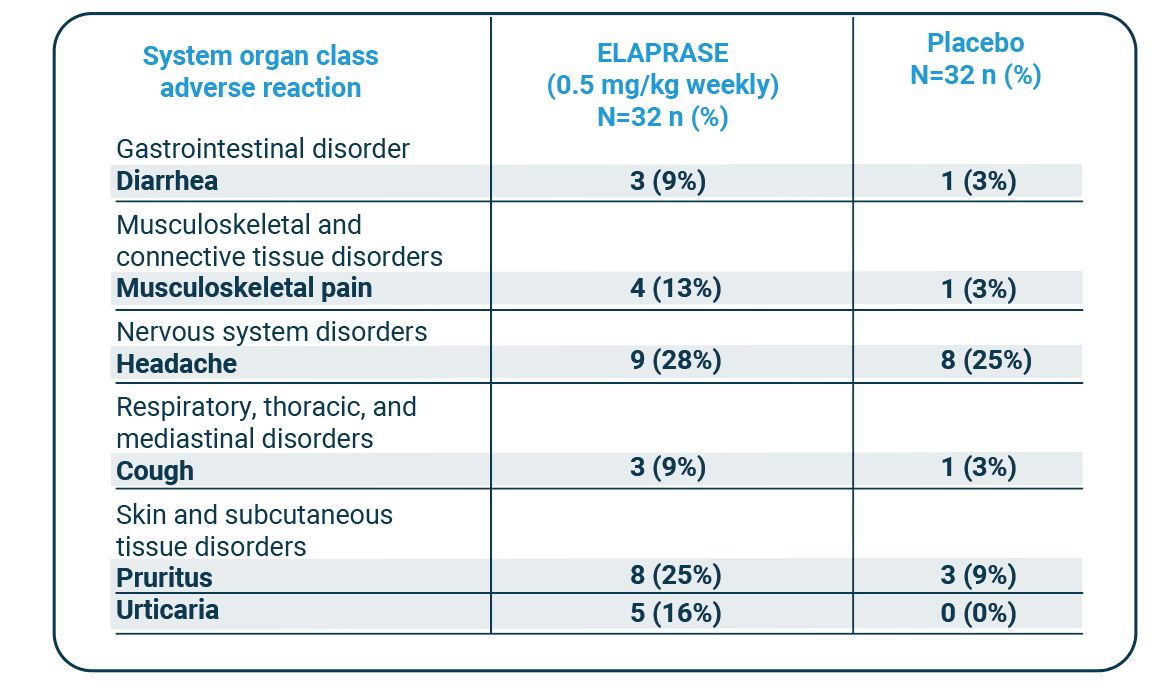
Additional adverse reactions that occurred in at least three patients (≥9%) in the ELAPRASE every-other-week group with a higher incidence than in the placebo group included: rash (19%), flushing (16%), fatigue (13%), tachycardia (9%), and chills (9%).1
Please see the ELAPRASE Important Safety Information here.
Pivotal trial primary endpoint
The composite primary endpoint differed statistically significantly between the three groups, and the difference was greatest between the placebo group and the once-weekly treatment group (once-weekly ELAPRASE vs. placebo, p=0.0049).
The changes from baseline to Week 53 in %-predicted FVC were not statistically significant.1,2
The results are displayed below:
| ELAPRASE weekly n=32* |
Placebo N=32* |
ELAPRASE Once weekly - Placebo |
|||||
|---|---|---|---|---|---|---|---|
| Baseline | Week 53 | Change† | Baseline | Week 53 | Change† | Difference in change | |
| Results from the 6-minute walk test (Meters) | |||||||
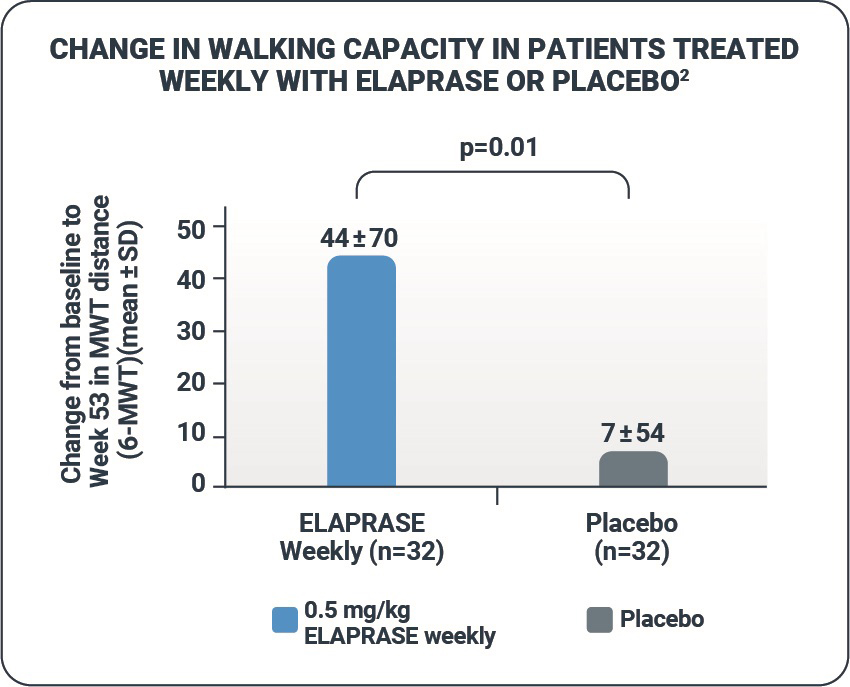
| Mean ± SD |
392 ± 108 |
436 ± 138 |
44 ± 70 |
393 ± 106 |
400 ± 106 |
7 ± 54 |
37 ± 16‡ 35 ± 14◊ (p=0.01) |
| Median | 397 | 429 | 31 | 403 | 412 | -4 | |
| Percentiles (25th, 75th) |
316, 488 |
365, 536 |
0, 94 |
341, 469 |
361, 460 |
-30, 31 |
|
| Results from the forced vital capacity test (% of predicted) | |||||||
| Mean ± SD |
55.3 ± 15.9 |
58.7 ± 19.3 |
3.4 ± 10.0 |
55.6 ± 12.3 |
56.3 ± 15.7 |
0.8 ± 9.6 |
2.7 ± 2.5‡ 4.3 ± 2.3◊ (p=0.07) |
| Median | 54.9 | 59.2 | 2.1 | 57.4 | 54.6 | −2.5 | |
| Percentiles (25th, 75th) |
43.6, 69.3 |
44.4, 70.7 |
−0.8, 9.5 |
46.9, 64.4 |
43.8, 67.5 |
−5.4, 5.0 |
|
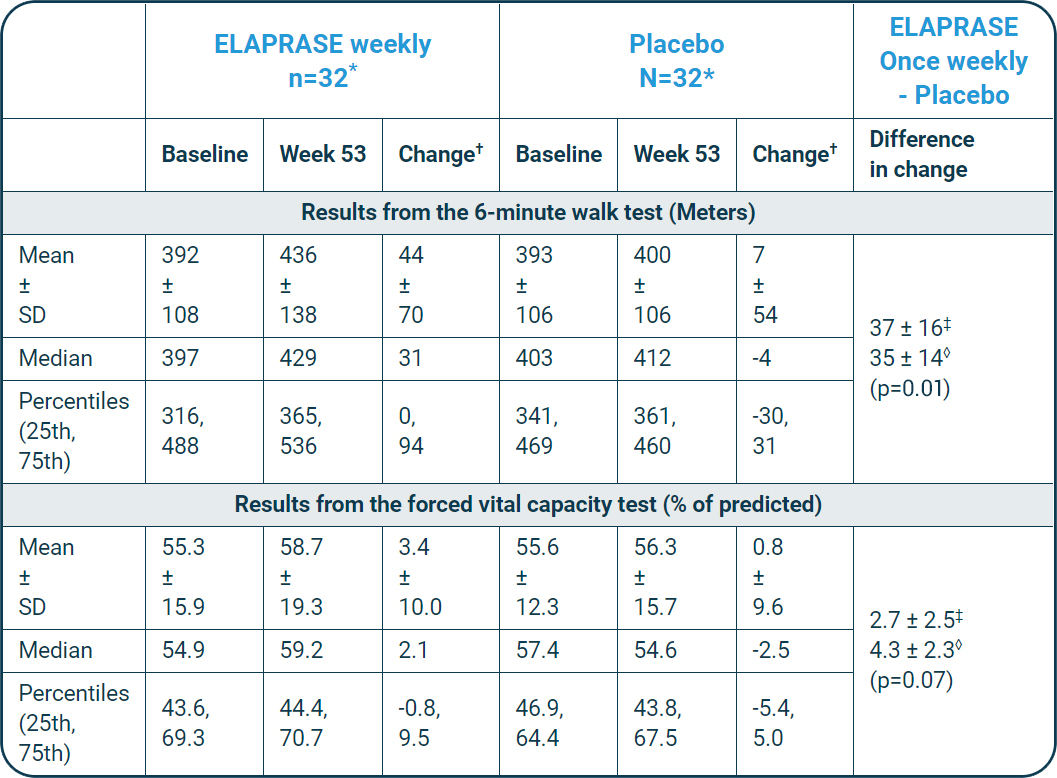
†Change, calculated as Week 53 minus baseline.
‡Observed mean ± SE.
◊ANCOVA model-based mean ± SE, adjusted for baseline disease severity, region, and age.
Read the ELAPRASE Important Safety Information or see below for more clinical trial data.
Pivotal trial secondary endpoints
Walking capacity
- ELAPRASE was shown to improve the distance walked in 6 minutes by a mean distance of 37 meters compared with placebo (p=0.01).1,2
- The 6-MWT is a self-paced assessment that reflects the pace and ability to carry out daily physical activities.3
Pulmonary function
- The changes from baseline to Week 53 in %-FVC were not statistically significant.1,2
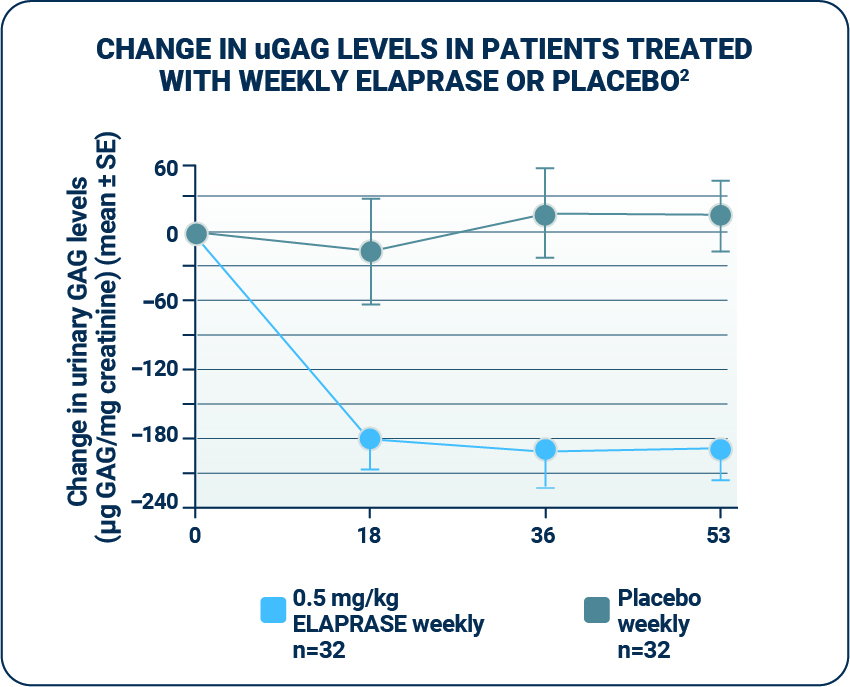
Urinary GAG levels
-
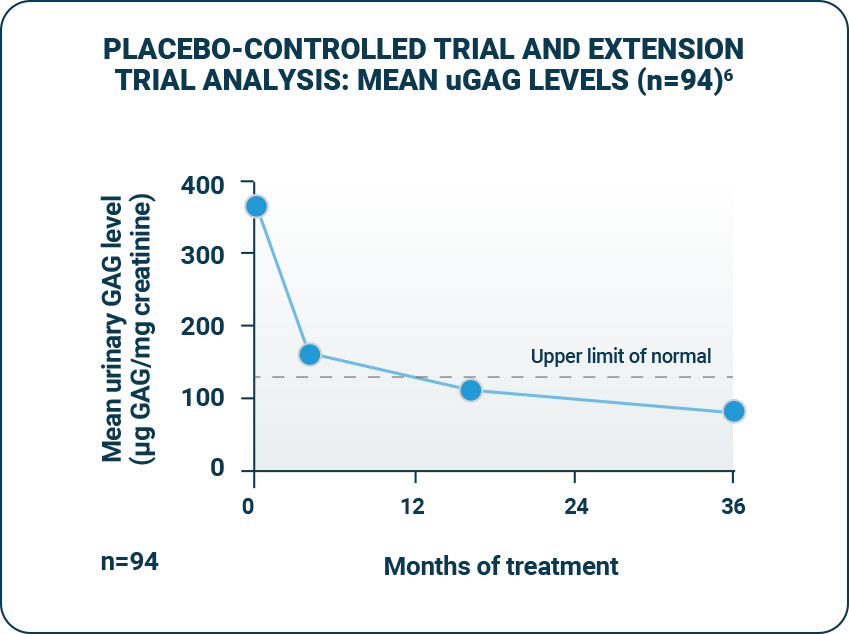
ELAPRASE was shown to reduce mean urinary GAG (uGAG) levels.1
- - uGAG levels were elevated in all patients at baseline; following 53 weeks of treatment, 50% of ELAPRASE-treated patients had uGAG levels below the upper limit of normal and 50% still remained above the upper limit of normal.1
- - Patients who tested positive for anti-idursulfase antibodies experienced a less-pronounced decrease in urinary GAG levels. The responsiveness of urinary GAG levels to dosage alterations of ELAPRASE is unknown.1,2
- uGAG levels represent the excretion of excess GAGs; therefore, a reduction in uGAG levels is a biochemical indication of ELAPRASE enzyme activity.4,5 However, the relationship of uGAG levels to other measures of clinical response has not been established.1
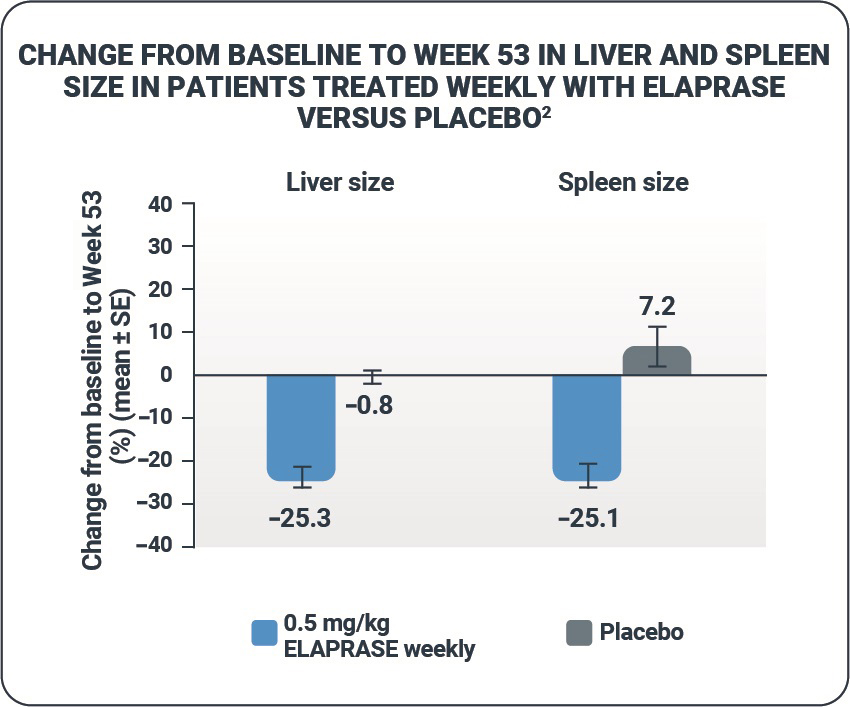
Liver volume and spleen volume
- ELAPRASE was shown to reduce mean liver and spleen volumes from baseline to Week 53 in the ELAPRASE once-weekly group.1,2
- There were essentially no changes in liver and spleen volumes in the placebo group.1,2
Extension trial design
- The extension trial was designed to evaluate the efficacy and safety of extended weekly treatment with ELAPRASE. The study ran for 24 months after the initial 53-week period.
- Patients in the pivotal trial were eligible to continue treatment in the open-label extension trial; 94 of 96 participants continued into the extension trial. All patients in the extension trial received ELAPRASE 0.5 mg/kg once weekly for 24 months.1
- The co-primary endpoints were 6-MWT and %-FVC.6
Extension trial safety profile
- No new serious adverse reactions were reported during the extension study.1
- Approximately half (53%) of patients experienced hypersensitivity reactions during the 24-month extension trial.
In addition to the frequently experienced adverse reactions (see Table 1) in the ELAPRASE once-weekly group in the pivotal trial (diarrhea [9%], musculoskeletal pain [13%], headache [28%], cough [9%], pruritus [25%], and urticaria [16%]), common hypersensitivity reactions occurring in at least five patients (≥5%) in the extension trial included:1
- Rash (23%)
- Pyrexia (9%)
- Flushing (7%)
- Erythema (7%)
- Nausea (5%)
- Dizziness (5%)
- Vomiting (5%)
- Hypotension (5%)
Read more about the ELAPRASE Important Safety Information here.
Extension trial data
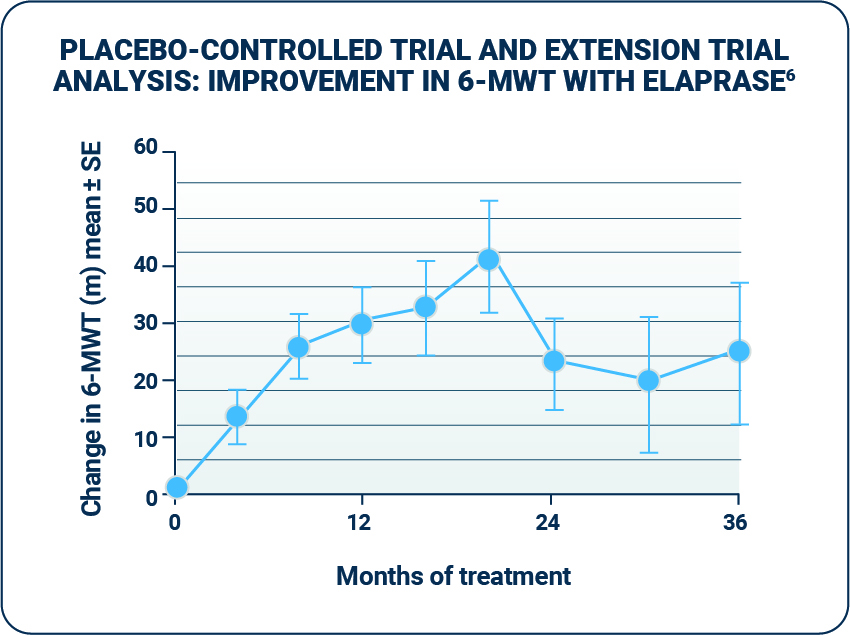
Walking capacity
- Patients treated with ELAPRASE demonstrated improvement in distance walked in the 6-MWT in both the placebo-controlled and extension trial.1
Pulmonary function
- There was no change in mean %-FVC in all Hunter syndrome patients after 6 months of treatment in the extension trial, followed by a slight decrease for the remainder of the 24-month period.1
- The %-FVC measure is affected by patient height; MPS II patients do not have normal growth rates, and so the interpretation of these data is not straightforward.6
- The long-term effect of ELAPRASE on pulmonary function in Hunter syndrome patients is unclear.1
Urinary GAG levels and liver/spleen volumes
- Reductions in uGAG levels displayed during the placebo-controlled trial were sustained during the extension trial.1
- Among patients treated with weekly ELAPRASE, no further reduction was observed during the extension trial.1
- The persistence of reduced uGAG levels did not correlate with the long-term effect demonstrated by the 6-MWT distance or %-FVC.1
- Reductions in liver and spleen volumes observed during the placebo-controlled trial were sustained during the extension trial.6
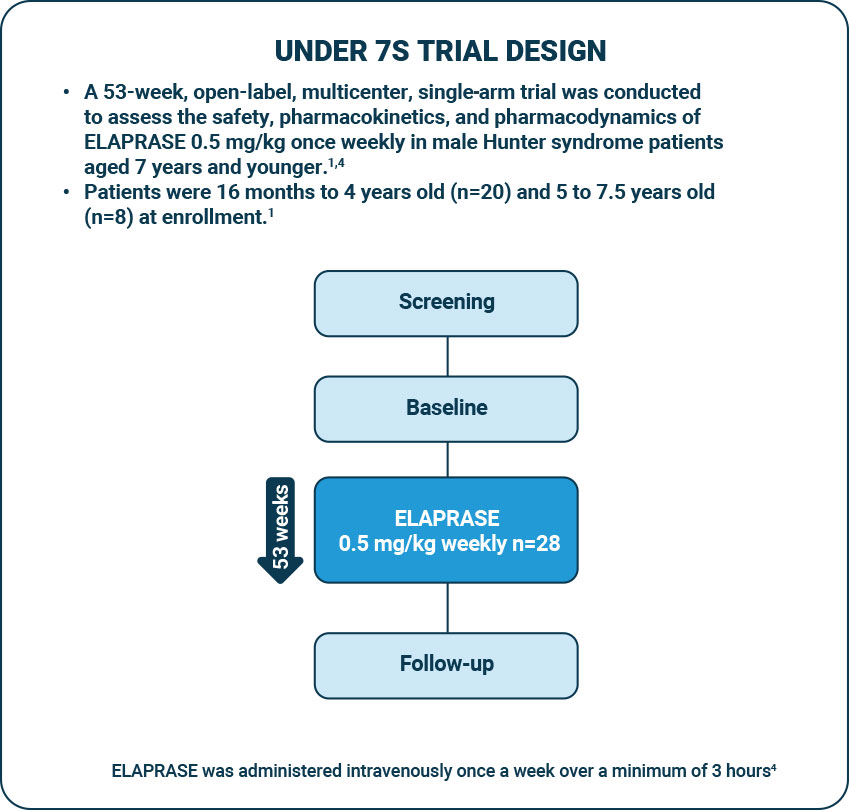
Under 7s trial design
Under 7s Safety profile
- Patients aged 16 months to 7.5 years experienced similar adverse reactions to those observed in clinical trials in patients aged ≥5 years. The most common adverse reactions following ELAPRASE treatment were hypersensitivity reactions (57%).1
- A higher incidence of the following common hypersensitivity reactions was reported in this younger age group: pyrexia (36%), rash (32%), and vomiting (14%).1
- The most common serious adverse reactions occurring in at least three patients (≥10%) included bronchopneumonia/pneumonia (18%), ear infection (11%), and pyrexia (11%).1
- Patients with complete gene deletion or large gene rearrangement mutations are more likely to develop antibodies, including neutralizing antibodies, and to experience hypersensitivity reactions and serious adverse events, compared with patients with missense mutations with ELAPRASE administration.1
Under 7s trial outcomes
In patients who remained antibody-negative:1
- A reduction in urinary GAG levels was observed, similar to the uGAG reduction seen in the pivotal trial.
- A reduction in spleen volume was observed, similar to the spleen volume reduction seen in the pivotal trial.
In patients who were persistently antibody-positive, the presence of anti-idursulfase antibody was associated with reduced systemic exposure of idursulfase and a less pronounced decrease in uGAG levels.1
In contrast, no apparent differences in pharmacokinetic parameter values between Week 1 and Week 27 were observed in the placebo-controlled trial among patients aged ≥5 years old receiving 0.5 mg/kg ELAPRASE (n=10), regardless of their antibody status.1
Read the ELAPRASE Important Safety Information here.
WARNING: RISK OF ANAPHYLAXIS
Life-threatening anaphylactic reactions have occurred in some patients during and up to 24 hours after ELAPRASE infusions. Anaphylaxis, presenting as respiratory distress, hypoxia, hypotension, urticaria and/or angioedema of throat or tongue have been reported to occur during and after ELAPRASE infusions, regardless of duration of the course of treatment. Closely observe patients during and after ELAPRASE administration and be prepared to manage anaphylaxis.